Predicting and Visualizing the Secondary Structure of RNA Sequences
This example illustrates how to use the rnafold and rnaplot functions to predict and plot the secondary structure of an RNA sequence.
Introduction
RNA plays an important role in the cell, both as genetic information carrier (mRNA) and as functional element (tRNA, rRNA). Because the function of an RNA sequence is largely associated with its structure, predicting the RNA structure from its sequence has become increasingly important. Because base pairing and base stacking represent the majority of the free energy contribution to folding, a good estimation of secondary structure can be very helpful not only in the interpretation of the function and reactivity, but also in the analysis of the tertiary structure of the RNA molecule.
RNA Secondary Structure Prediction Using Nearest-Neighbor Thermodynamic Model
The secondary structure of an RNA sequence is determined by the interaction between its bases, including hydrogen bonding and base stacking. One of the many methods for RNA secondary structure prediction uses the nearest-neighbor model and minimizes the total free energy associated with an RNA structure. The minimum free energy is estimated by summing individual energy contributions from base pair stacking, hairpins, bulges, internal loops and multi-branch loops. The energy contributions of these elements are sequence- and length-dependent and have been experimentally determined [1]. The rnafold function uses the nearest-neighbor thermodynamic model to predict the minimum free-energy secondary structure of an RNA sequence. More specifically, the algorithm implemented in rnafold uses dynamic programming to compute the energy contributions of all possible elementary substructures and then predicts the secondary structure by considering the combination of elementary substructures whose total free energy is minimum. In this computation, the contribution of coaxially stacked helices is not accounted for, and the formation of pseudoknots (non-nested structural elements) is forbidden.
Secondary Structure of Transfer RNA Phenylalanine
tRNAs are small molecules (73-93 nucleotides) that during translation transfer specific amino acids to the growing polypeptide chain at the ribosomal site. Although at least one tRNA molecule exists for each amino acid type, both secondary and tertiary structures are well conserved among the various tRNA types, most likely because of the necessity of maintaining reliable interaction with the ribosome. We consider the following tRNA-Phe sequence from Saccharomyces cerevisiae and predict the minimum free-energy secondary structure using the function rnafold.
% === Predict secondary structure in bracket notation phe_seq = 'GCGGAUUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUCUGGAGGUCCUGUGUUCGAUCCACAGAAUUCGCACCA'; phe_str = rnafold(phe_seq)
phe_str =
'(((((((..((((........)))).(((((.......))))).....(((((.......))))))))))))....'
In the bracket notation, each dot represents an unpaired base, while a pair of equally nested, opening and closing brackets represents a base pair. Alternative representations of RNA secondary structures can be drawn using the function rnaplot. For example, the structure predicted above can be displayed as a rooted tree, where leaf nodes correspond to unpaired residues and internal nodes (except the root) correspond to base pairs. You can display the position and type of each residue by clicking on the corresponding node.
% === Plot RNA secondary structure as tree rnaplot(phe_str, 'seq', phe_seq, 'format', 'tree');

The tRNA secondary structure is commonly represented in a diagram plot and resembles a clover leaf. It displays four base-paired stems (or "arms") and three loops. Each of the four stems has been extensively studied and characterized: acceptor stem (positions 1-7 and 66-72), D-stem (positions 10-13 and 22-25), anticodon stem (positions 27-31 and 39-43) and T-stem (positions 49-53 and 61-65). We can draw the tRNA secondary structure as a two-dimensional plot where each residue is identified by a dot and the backbone and the hydrogen bonds are represented as lines between the dots. The stems consist of consecutive stretches of base paired residues (blue dots), while the loops are formed by unpaired residues (red dots).
% === Plot the secondary structure using the dot diagram representation rnaplot(phe_str, 'seq', phe_seq, 'format', 'dot'); text(500, 200, 'T-stem'); text(100, 600, 'Anticodon stem'); text(550, 650, 'D-stem stem'); text(700, 400, 'Acceptor stem');

While all the stems are important for a proper three-dimensional folding of the molecule and successful interplay with ribosome and tRNA synthetases, the acceptor stem and the anticodon stem are particularly interesting because they include the attachment site and the anticodon triplet. The attachment site (positions 74-76) occurs at the 3' end of the RNA chains and consists of the sequence C-C-A in all amino acid acceptor stems. The anticodon triplet consists of 3 bases that pair with a complementary codon in the messenger RNA. In the case of Phe-tRNA, the anticodon sequence A-A-G (positions 34-36) pairs with the mRNA codon U-U-C, encoding the amino acid phenylalanine. We can redraw the structure and highlight these regions in the acceptor stem and anticodon stem by using the selection property:
aag_pos = 34:36; cca_pos = 74:76; rnaplot(phe_str, 'sequence', phe_seq, 'format', 'diagram', ... 'selection', [aag_pos, cca_pos]);

The segregation of the sequence into four separate stems is better appreciated by displaying the structure as graph plot. Each residue is represented on the abscissa and semi-elliptical lines connect bases that pair with each other. The lack of pseudoknots in the secondary structure is reflected by the absence of intersecting lines. This is expected in tRNA secondary structures and anticipated because the dynamic programming method used does not allow pseudoknots.
rnaplot(phe_str, 'sequence', phe_seq, 'format', 'graph');
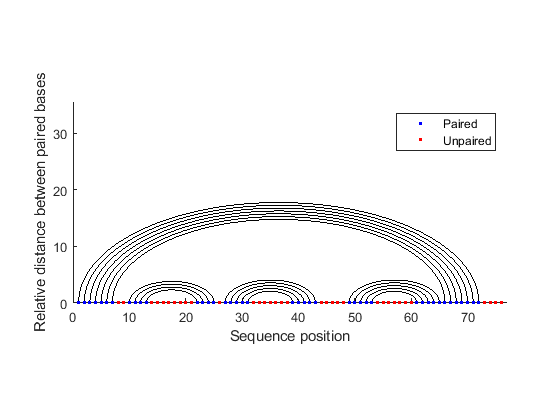
Similar observations can be drawn by displaying the secondary structure as a circle, where each base is represented by a dot on the circumference of a circle of arbitrary size, and bases that pair with each other are connected by lines. The lines are visually clustered into four distinct groups, separated by stretched of unpaired residues. We can hide the unpaired residues by using H.Unpaired, the handle returned with the colorby property set to state.
[ha, H] = rnaplot(phe_str, 'sequence', phe_seq, 'format', 'circle', ... 'colorby', 'state'); H.Unpaired.Visible = 'off'; legend off;

As you can see, the outputs of the rnaplot function include a MATLAB® structure H consisting of handles that can be used to change the aspect properties of various residue subsets. For example, if you set the color scheme using the colorby property set to residue, the dots are colored according to the residue type, and you can change their property using the appropriate handle.
[ha, H] = rnaplot(phe_str, 'sequence', phe_seq, 'format', 'circle', 'colorby', 'residue')
ha =
Axes (Bioinfo:rnaplot:circle) with properties:
XLim: [-1 1]
YLim: [-1 1.1000]
XScale: 'linear'
YScale: 'linear'
GridLineStyle: '-'
Position: [0.1124 0.1100 0.6703 0.8150]
Units: 'normalized'
Use GET to show all properties
H =
struct with fields:
A: [1×1 Line]
C: [1×1 Line]
G: [1×1 Line]
U: [1×1 Line]
Selected: [0×1 Line]

H.G.Color = [0.5 0.5 0.5]; H.G.Marker = '*'; H.C.Color = [0.5 0.5 0.5]; H.C.Marker = '+';

Conservation of Transfer RNA Phenylalanine
Despite some differences in their primary sequences, tRNAs molecules present a secondary structure pattern that is well conserved across the three phylogenetic domains. Consider the structure of the tRNA-Phe of one representative organism for each phylogenetic domain: Saccharomyces cerevisiae for the Eukaryotes, Haloarcula marismortui for the Archaea, and Thermus thermophilus for the Bacteria. Then predict and plot their secondary structures using the mountain plot representation.
yeast = 'GCGGACUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUCUGGAGGUCCUGUGUUCGAUCCACAGAGUUCGCACCA'; halma = 'GCCGCCUUAGCUCAGACUGGGAGAGCACUCGACUGAAGAUCGAGCUGUCCCCGGUUCAAAUCCGGGAGGCGGCACCA'; theth = 'GCCGAGGUAGCUCAGUUGGUAGAGCAUGCGACUGAAAAUCGCAGUGUCGGCGGUUCGAUUCCGCCCCUCGGCACCA'; yeast_str = rnafold(yeast); theth_str = rnafold(theth); halma_str = rnafold(halma); h1 = rnaplot(yeast_str, 'sequence', yeast, 'format', 'mountain'); title(h1, 'tRNA-Phe Saccharomyces cerevisiae'); legend hide; h2 = rnaplot(halma_str, 'sequence', halma, 'format', 'mountain'); title(h2, 'tRNA-Phe Haloarcula marismortui'); legend hide; h3 = rnaplot(theth_str, 'sequence', theth, 'format', 'mountain'); title(h3, 'tRNA-Phe Thermus thermophilus'); legend hide;



The similarity among the resulting structures is striking, the only difference being one extra residue in the D-loop of Haloarcula marismortui, displayed in the first flat slope in the mountain plot.
The G-U Wobble Base Pair
Besides the Watson-Crick base pairs (A-U, G-C), virtually every class of functional RNA presents G-U wobble base pairs. G-U pairs have an array of distinctive chemical, structural and conformational properties: they have high affinity for metal ions, they are almost thermodynamically as stable as Watson-Crick base pairs, and they present conformational flexibility to different environments. The wobble pair at the third position of the acceptor helix of tRNA is very highly conserved in almost all organisms. This conservation suggests that the G-U pair possesses unique features that can hardly be duplicated by other pairs. You can observe the base pair type distribution on the secondary structure diagram by coloring the base pairs according to their type.
rnaplot(yeast_str, 'sequence', yeast, 'format', 'diagram', 'colorby', 'pair');

References
[1] Matthews, D., Sabina, J., Zuker, M., and Turner, D. "Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure", Journal of Molecular Biology, 288(5):911-40, 1999.